Абдоминальная
с развитием асептического перитонита, проявляющегося выраженным болевым синдромом с картиной «острого живота»
Семейная средиземноморская лихорадка (Familial Mediterranean Fever, FMF) — аутовоспалительное заболевание, характеризующееся периодически повторяющимися приступами лихорадки в сочетании с асептическим перитонитом, иногда плевритом, поражением кожи, артритом и редко перикардитом [1]. FMF дебютирует, как правило, до 20 лет.
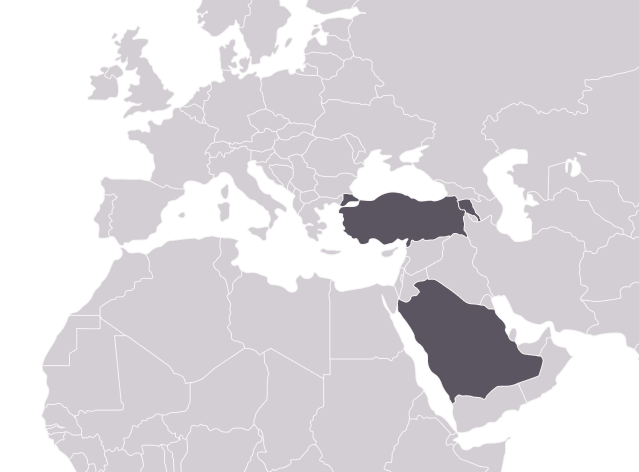
Среди всех аутовоспалительных заболеваний для FMF в наибольшей степени характерна этническая предрасположенность. Наиболее подвержены заболеванию представители четырех этносов: евреи-сефарды (североафриканские, неашкеназские евреи), турки, арабы и армяне [1]. Знание этнической принадлежности пациента оказывает существенную помощь при установлении диагноза.
По мере изучения FMF стало понятно, что распространенность FMF не ограничивается указанными выше популяциями. Благодаря миграции населения в ХХ веке, заболевание распространилось по всему миру: США, Европа, Россия, Япония, Южная Америка и др. [2]. Таким образом, заболевание было выявлено у представителей достаточно отдаленных этнических групп, что предостерегает от постановки диагноза исключительно на основе родословной.
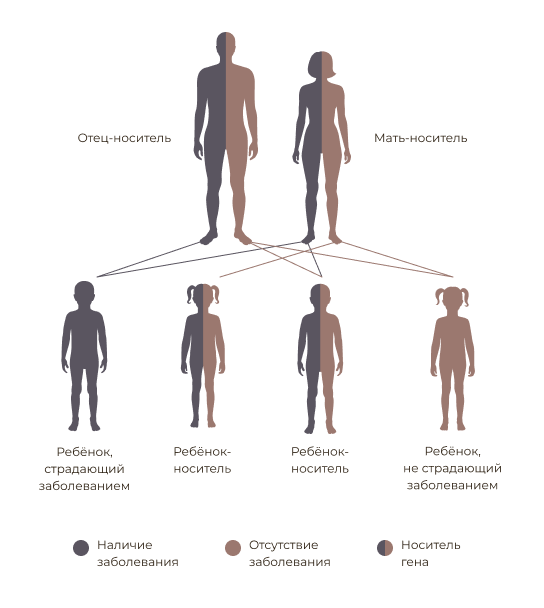
Этиологическим фактором FMF выступает мутация гена MEFV, локализованного на коротком плече 16-й пары хромосом. Тип наследования — аутосомно-рецессивный, соответственно, если оба родителя являются здоровыми гетерозиготами, вероятность развития заболевания у их потомства составляет 25%. Вместе с тем имеются данные о возможности аутосомно-доминантного типа наследования при наличии мутации с высокой пенетрантностью [3].
Мутации, ассоциированные с FMF, являются мутациями усиления функции, то есть они придают белку новую или повышенную активность с эффектом дозы аномального гена (т. е. увеличение количества копий аномального гена дает больший эффект). Ген MEFV в норме кодирует белок (так называемый пирин), который экспрессируется в циркулирующих нейтрофилах [4].
Пирин представляет собой неотъемлемый структурный компонент врожденной иммунной системы и участвует в защитных реакциях от бактериальных токсинов, которые деполимеризуют актин и активируют инфламмасомы. Бактерии, которые производят подобные токсины, включают Clostridium difficile, Burkholderia cenocepacia, а также Vibrio cholerae [4]. Предполагается, что пирин притупляет воспалительный ответ, возможно, путем ингибирования хемотаксиса и активации нейтрофилов. Генные мутации приводят к появлению измененных молекул пирина, которые не ингибируют активацию инфламмасом.
Нарушение функции инфламмасом ведет к спонтанному запуску воспалительного каскада и гиперактивации системы врожденного иммунитета без видимой причины (в отсутствие инфекции или аутоиммунной реакции), основная патогенетическая роль в этом процессе принадлежит провоспалительному цитокину ИЛ-1β [4, 5].
Интерлейкин-1β активирует клетки врожденного иммунитета (нейтрофилы, моноциты; Т-лимфоциты; остеокласты), индуцирует экспрессию коллагеназ, разрушающих суставной хрящ, стимулирует выработку простагландина E2 в гипоталамусе, что приводит к развитию гипертермии центрального генеза. Воздействуя на эндотелий сосудов, ИЛ-1β способствует развитию сыпи и стимулирует атерогенез в кровеносных сосудах, а также активирует синтез острофазовых белков в печени [5].
Клинические последствия гиперпродукции ИЛ-1β — спонтанные приступы системного воспаления с увеличением количества лейкоцитов за счет нейтрофилов и острофазовых показателей крови.
Лихорадка: температура свыше 38 °С в течение 12–72 часов во время типичного приступа
Сыпь: болезненное, четко очерченное, сильное покраснение кожи примерно у 40% пациентов — часто в области ступней или нижней части ног
Существенный риск приобретенного амилоидоза — у 50% пациентов
У отдельных пациентов наблюдаются:

Воспаление сустава (моноартрит), например коленного, голеностопного или запястного
FMF характеризуется периодическими повторяющимися атаками системного воспаления продолжительностью 6–72 ч, проявляющегося фебрильной лихорадкой 38–40,5 °С и симптомами воспаления органов и систем [6, 7].
Лихорадка: температура свыше 38 °С в течение 12–72 часов во время типичного приступа
Сыпь: болезненное, чётко очерченное, сильное покраснение кожи примерно у 40% пациентов — часто в области ступнёй или нижней части ног
Сыпь: болезненное, чётко очерченное, сильное покраснение кожи примерно у 40% пациентов — часто в области ступнёй или нижней части ног
Существенный риск приобретенного амилоидоза у 50% пациентов
У отдельных пациентов наблюдаются:
FMF характеризуется периодическими повторяющимися атаками системного воспаления продолжительностью 6–72 ч, проявляющегося фебрильной лихорадкой 38–40,5 °С и симптомами воспаления органов и систем [6, 7].
с развитием асептического перитонита, проявляющегося выраженным болевым синдромом с картиной «острого живота»
с развитием артритов, главным образом суставов нижних конечностей
с развитием асептического плеврита, в большинстве случаев одностороннего
Основное проявление — рожеподобная эритема, как правило на голенях, над голеностопным суставом, в котором имеются признаки артрита, и на тыльной поверхности стоп
Частота атак FMF различна, что отражает ее тяжесть: от 1–2 раз в неделю до нескольких в год. Во время атаки возможны любые сочетания фебрильной лихорадки с проявлениями воспаления со стороны указанных систем. Редко атаки могут протекать в виде изолированной лихорадки. Практически всегда фебрильные атаки сопровождаются значительным повышением уровня острофазовых маркеров: СОЭ, СРБ, сывороточного амилоида А (SAA) [8, 9].
Значения острофазовых маркеров существенно снижаются вне атаки, но примерно у 63% пациентов сохраняется повышенный уровень того или иного острофазового показателя в промежутке между атаками. Ключевым и наиболее чувствительным маркером воспаления, в качестве оценки активности заболевания, эффективности лечения и риска развития амилоидоза, в настоящее время служит сывороточный амилоид А (SAA) [3, 10].
Амилоидоз почек — основное осложнение, которое определяет прогноз заболевания и приводит к развитию хронической почечной недостаточности. Амилоидоз почек до 60% случаев является причиной смертности больных с FMF [14]. Частота амилоидоза составляет 25–30% [1] и может зависеть от этнического происхождения пациента [15]. Так, у нелеченых пациентов турецкого происхождения частота амилоидоза достигает 60%, у евреев-сефардов — 75% [15].
Общая популяция
Турки
Евреи-сефарды
Другим осложнением, имеющим существенно меньшее значение для прогноза заболевания, является спаечная болезнь.
Клиническими последствиями воспаления при FMF могут быть нормоцитарно-нормохромная анемия, спленомегалия, задержка роста у детей, снижение плотности костей, мужское и женское бесплодие, преждевременные роды, повышенный риск сердечно-сосудистых заболеваний [11].
Заподозрить диагноз могут помочь клинические диагностические / классификационные критерии EUROFEVER [15].
Диагностические критерииПодтверждает диагноз обнаружение мутаций гена MEFV. Генетическое тестирование проводят в специализированных лабораториях, имеющих опыт диагностики мАВЗ [1].
Генетический проектГенетическое тестирование используется для подтверждения, но не для исключения диагноза периодической болезни. Наиболее часто встречаются 5 патогенных мутаций гена MEFV: M694V, M694I, M680I, V726A, E148Q. Наибольшее число мутаций (до 38%) приходится на 10-й экзон. Мутации M694V приводят к более тяжелому течению заболевания с ранним началом, частому поражению суставов, высокому уровню амилоидоза при отсутствии лечения. Пациенты, гомозиготные по мутации M694V, в 6 раз чаще страдают амилоидозом по сравнению с другими генотипами. Мутации обнаруживаются также в экзонах [2, 3, 16, 17, 18].
Следует иметь в виду, что у некоторых пациентов с очевидными фенотипическими признаками FMF выявляется только один мутантный ген, а иногда отсутствуют явные мутации в гене MEFV. Приблизительно у 10–20% пациентов, которые отвечают диагностическим критериям FMF, мутации в гене MEFV не выявляются, что предполагает наличие эпигенетических факторов и факторов внешней среды, способствующих патогенезу заболевания [19].

Приблизительно у 10–20% пациентов, которые отвечают диагностическим критериям FMF, мутации в гене MEFV не выявляются.
Неспецифические признаки включают: повышение уровня лейкоцитов в крови с преобладанием нейтрофилов, СОЭ, С-реактивного белка и фибриногена. Экскреция с мочой > 0,5 г белка за сутки предполагает формирование амилоидоза почек [19].
Цель лечения FMF — достижение полного контроля над неспровоцированными приступами воспаления и минимизация субклинического воспаления в межприступный период [22].
В настоящее время основное лечение FMF заключается в применении колхицина. Колхицин предотвращает атаки заболевания, снижает их частоту и тяжесть. Также важным фактором служит значительное уменьшение риска развития амилоидоза при применении адекватных доз препарата [23].
Сегодня колхицин — единственный препарат, эффективность которого в отношении предотвращения амилоидоза доказана. В соответствии с рекомендациями EULAR (2016) колхицин должен быть назначен сразу же после установления диагноза FMF [23].
Колхицин эффективен у большинства, но далеко не у всех пациентов с FMF. У 20% пациентов с FMF на терапии колхицином сохраняются воспалительные атаки и скрытое воспаление между приступами. Почему это происходит? Более 40% пациентов не соблюдают рекомендации по лечению, 2–5% отмечают непереносимость и серьезные побочные эффекты терапии колхицином (тяжелая диарея, невропатия, рабдомиолиз и подавление активности костного мозга) [23]. Кроме того, существует понятие «колхицинрезистентности», когда пациент с FMF недостаточно отвечает на терапию максимально допустимой дозой колхицина. Колхицинрезистентность диагностируется при сохранении ≥ 1 атаки в месяц у комплаентных пациентов (получающих максимальную дозу колхицина ≥ 6 мес.) или при стойком повышении уровня СРБ/SАА в период между приступами [22, 25, 26].
Доля пациентов с истинной колхицинрезистентностью достигает 15%. Таким пациентам показано назначение генно-инженерных биологических препаратов (ГИБП) [25].

Изучение молекулярно-биологических основ развития заболевания позволило выбрать эффективные мишени для терапии. Пациенты с любым генетическим паттерном и разнообразием клинических проявлений хорошо отвечают на лечение генно-инженерными биологическими препаратами (ГИБП) [27].
Выявление АА-амилоидоза требует рассмотрения возможности назначения биологической терапии вне зависимости от наличия колхицинрезистентности у пациентов с FMF [25].
FMF является самым распространенным моногенным аутовоспалительным заболеванием и имеет отчетливую этническую предрасположенность. В России проживает немало представителей этнических групп (армяне, азербайджанцы, народы Северного Кавказа), в которых отмечается высокая распространенность мутации гена MEFV и, соответственно, FMF. Благодаря внедрению в клиническую практику молекулярной генетической диагностики выявляемость FMF увеличивается.
Тяжесть и прогноз заболевания определяются не только атаками системного воспаления, но и высоким риском развития вторичного АА-амилоидоза, который может привести к развитию хронической почечной недостаточности и гибели пациента. Патогенетическая лекарственная терапия ГИБП позволяет достичь значительного прогресса в ведении таких пациентов с улучшением прогноза, предотвращением развития жизнеугрожающих осложнений и улучшением качества жизни [1].



Получить информацию о генетической диагностике аутовоспалительных заболеваний можно по номеру горячей линии 8 800 100-15-92 в рабочее время с 8:00 до 18:00 (мск).
FCAS
12–24 ч.
MWS
2–3 дня
NOMID
Непрерывная с периодическим усилением симптомов
FCAS
Конъюнктивит
MWS
NOMID
Непрерывная с периодическим усилением симптомов
FCAS
Холодозависимая нейтрофильная уртикарная сыпь (крапивница)
MWS
Нейтрофильная уртикарная сыпь (крапивница)
NOMID
Нейтрофильная уртикарная сыпь (крапивница)
FCAS
Не характерно
MWS
NOMID
FCAS
Головная боль
MWS
NOMID
FCAS
MWS
NOMID
Встречаются редко. Конъюнктивит, нумулярный кератит, приводящий к значительной потере зрения. Клинически проявляется светобоязнью, болью и резью в глазах, покраснением глаз [9].
Встречаются редко. Конъюнктивит, нумулярный кератит, приводящий к значительной потере зрения. Клинически проявляется светобоязнью, болью и резью в глазах, покраснением глаз [9].
Одно из наиболее характерных проявлений. Чаще увеличиваются шейные лимфатические узлы. Увеличение обычно двустороннее, иногда достигает значительных размеров, меняя контуры шеи.
Обязательный симптом. Достигает фебрильных значений, часто сопровождается ознобами. Продолжительность эпизода не превышает 1 недели, обычно 3–5 дней. Интервалы между приступами от 2 до 8 недель [6], по некоторым данным — от 3 до 6 недель [7].
Весьма типичны боли в животе, нередко выраженного характера, сопровождающиеся тошнотой и рвотой, диареей.
Диарея встречается у 82% пациентов, стул может быть водянистым, изредка с примесью крови [7], однако ее эпизоды непродолжительны.
Наиболее типична головная боль во время атаки. Описаны редкие случаи более серьезных неврологических нарушений с задержкой психического развития различной степени тяжести, атаксией и эпилептиформными приступами [8].
Описаны случаи мембранозно-пролиферативного гломерулонефрита, проявлявшегося макрогематурией, выраженной протеинурией и артериальной гипертензией [11, 12].
Частое проявление. Артралгии, транзиторный неэрозивный артрит с поражением преимущественно коленных и голеностопных суставов.
Выраженное ухудшение самочувствия во время атаки, слабость, разбитость.
Наиболее типична головная боль во время атаки. Описаны редкие случаи более серьезных неврологических нарушений с задержкой психического развития различной степени тяжести, атаксией и эпилептиформными приступами [8].
Наиболее типична головная боль во время атаки. Описаны редкие случаи более серьезных неврологических нарушений с задержкой психического развития различной степени тяжести, атаксией и эпилептиформными приступами [8].
Ссылка ведет на ресурс, на который не распространяется наша политика по безопасности персональных данных. Компания "Новартис" не несет ответственность за содержание стороннего ресурса.