Гипер-IgD-синдром / синдром дефицита мевалонаткиназы. Общая информация
Гипер-IgD-синдром / синдром дефицита мевалонаткиназы (Hyper-Immunoglobulinemia D-syndrome / Mevalonate Kinasae Deficiency syndrome — HIDS/MKD) относят к группе моногенных аутовоспалительных заболеваний (мАВЗ) [1]. Синдром впервые описан в 1984 г. J.W.M. van der Meer и соавт. [2].
Распространенность
Частота заболевания ориентировочно составляет 1:1 000 000, но точно не определена [3]. Известно, что заболевание распространено среди датчан, голландцев, французов [1].
Этиология и патогенез

HIDS наследуется по аутосомно-рецессивному типу. Синдром обусловлен мутацией гена на длинном плече 12-й пары хромосом, который кодирует фермент мевалонаткиназу [1, 3].
Мевалонаткиназа принимает участие в синтезе холестерина и родственных соединений (стероидов, желчных кислот, компонентов клеточных мембран, липидных компонентов соединений липопротеидной природы) [5]. Патогенетические механизмы развития воспаления при мАВЗ, в частности HIDS, представлены ниже.
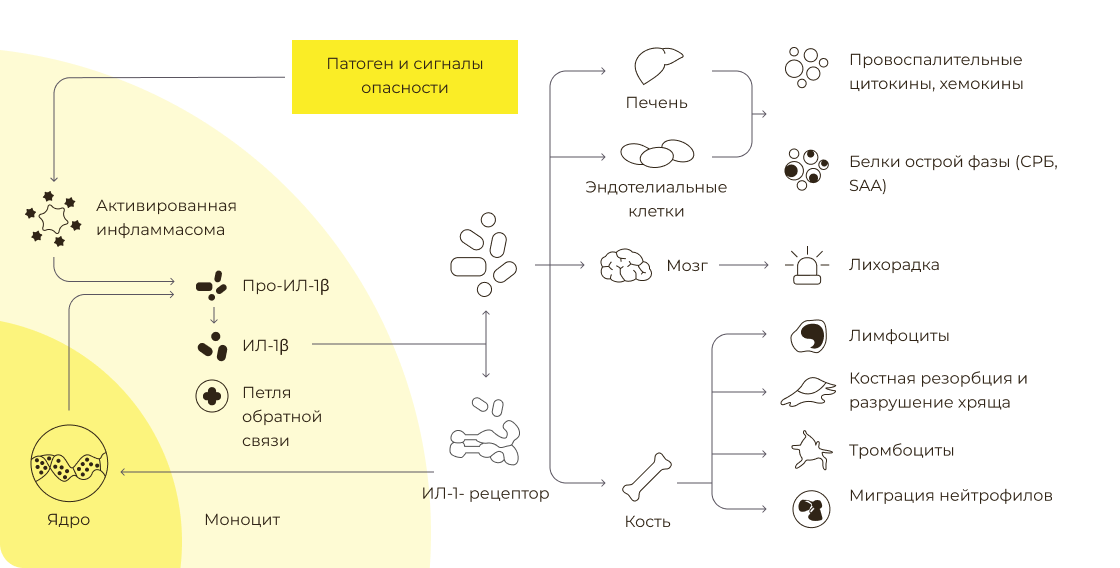
Патогенетические механизмы развития воспаления при мАВЗ:
Нажмите чтобы увеличить
Симптомы HIDS являются результатом недостаточного мевалонового фосфорилирования в биосинтезе стерола [3]. Сам по себе избыток мевалоната не способствует воспалительной атаке, она возникает из-за недостатка изопреноидов. Снижение геранилгеранил пирофосфата запускает активацию каспазы-1 за счет передачи сигналов Rac1. Каспаза-1, в свою очередь, активирует про-интерлейкин-1β (про-ИЛ-1β), а затем ИЛ-1β, который способствует развитию цитокинового шторма.
В этих процессах участвуют NLRP3-инфламмасомы — компонент врожденного иммунитета. Инфламмасомы активируют молекулярные паттерны, ассоциированные с повреждениями (damage associated molecular patterns — DAMP), патоген-ассоциированные молекулярные паттерны (pathogen-associated molecular patterns — PAMP), например липополисахариды или сигналы поврежденных клеток. Это может объяснить, почему в некоторых случаях инфекция, вакцина или стресс служат триггерами в развитии синдрома дефицита мевалонаткиназы [3].
Клиническая картина
Заболевание развивается в раннем возрасте, чаще в течение первых двух лет жизни. Дебют могут спровоцировать стрессовые факторы: инфекции, травмы, хирургические вмешательства, эмоциональные нагрузки, часто — вакцинация [5].
Клиническая картина включает следующие признаки [5]:
Гепатоспленомегалия
Сыпь
(пятнисто-папулезная, уртикарная, петехиально-пурпурная)
Головная боль
Шейная лимфаденопатия
Артралгии, артрит крупных суставов
Боль в животе, рвота, диарея
Оральные, генитальные язвы
Подробные характеристики симптомов приведены ниже:

Офтальмологические проявления
Лимфаденопатия
Лихорадка
Поражение желудочно-кишечного тракта
Поражение кожи и слизистых оболочек
Поражение почек
Поражение суставов
Общая интоксикация
Поражение нервной системы
Поражение нервной системы
Наиболее типична головная боль во время атаки. Описаны редкие случаи более серьезных неврологических нарушений с задержкой психического развития различной степени тяжести, атаксией и эпилептиформными приступами [8]
ДалееДиагностика
Заподозрить диагноз могут помочь клинические диагностические / классификационные критерии EUROFEVER [14].
Диагностические критерииПодтверждает диагноз обнаружение мутаций гена мевалонаткиназы. Генетическое тестирование проводят в специализированных лабораториях, имеющих опыт диагностики мАВЗ [1].
Генетический проектЛечение
Изучение молекулярно-биологических основ развития заболевания позволило выбрать эффективные мишени для терапии. Пациенты с любым генетическим паттерном и разнообразием клинических проявлений хорошо отвечают на лечение генно-инженерными биологическими препаратами (ГИБП) [15].
Доказательная база по лечению HIDS складывается в основном из данных ретроспективных исследований. Применение нестероидных противовоспалительных препаратов (НПВП) позволяет уменьшить симптомы во время обострений, но не купирует саму воспалительную атаку. Аналогичные данные получены и в отношении глюкокортикоидов [15–17].
Прогноз

Пациенты с HIDS имеют благоприятный прогноз. Амилоидоз регистрируют в менее 3% случаев. Заболевание может не проявлять себя в течение нескольких месяцев и даже лет. Артрит при этом заболевании не деструктивный и не приводит к инвалидизации [5].
Источники
- Салугина С.О., Федоров У.С., Агафонова Е.С. Моногенные аутовоспалительные заболевания детей и взрослых: что необходимо знать ревматологу. Научно-практическая ревматология. 2019;57(2):125–132. doi: 10.14412/1995–4484–2019–125–132.
- Van der Meer J.W.М., Vossen J.M., Radl J. et al. Hyperimmunoglobulinemia D and periodic fever: a new syndrome. Lancet 1984;1:1087–90.
- Козлова А.Л. и др. // Вопросы гематологии/онкологии и иммунопатологии в педиатрии, 2016, т. 15, № 1, с. 46–53.
- Drenth G., van der Meer J.W.М. Hereditary periodic fever. N Engl J Med 2001;345(24):1748–57.
- Кузьмина Н.Н., Салугина С.О., Федоров Е.С. Аутовоспалительные заболевания и синдромы у детей: Учеб.-метод. Пособие. — М: ИМА-ПРЕСС, 2012. — 104 с.
- Fietta P. Autoinflammatory disease: the hereditary periodic fever syndromes. Acta Biol Ateneo Parmense 2004;75:92–9.
- Lierl M. Periodic fever syndromes: a diagnostic challendge for the allergist. Allergy 2007;62:1349–58.
- Haas D., Hoffman F. Mevalonate kinase deficiencies: from mevalonic aciduria to hyperimmunoglobulinemia D syndrome. Orphanet J Rare Dis 2006;1:13.
- Kraus C.L., Culican S.M. Nummular keratopathy in patien with hyper IgD syndrome. Ped Rheumatol 2009;7:14.
- Arbrust S., Drenth J.P., Schroder C. et al. Tendonitis in variant hyperimmunoglobulinemia D and periodic fever syndrome – a rare disease with a new symptom. Eur J Pediatr 2005;164:391–4.
- D’Osualdo A., Picco P., Caroli F. et al. MVK mutation and assotiated clinical featers in Italian patients affected with autoinflammatory disorders and recurrent fever. Eur J Hum Gen. 2005;13:314–20.
- Nevyjel M., Pontillo A., Calligaris L. et al. Diagnostics and therapeutic insights in a severe case of mevalonate kinase deficiency. Pediatrics 2007;119:523–7.
- Drenth J.P., Guisset L., Grateau G. et al. Mutation in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. Nat Genet. 1999;22:178–81.
- Federici S., Sormani M., Ozen S. et al. Evidence-based provisional clinical classification criteria for autoinflammatory periodic fevers. Ann Rheum Dis. 2015;74:799-805.
- Goldbach-Mansky R. Blocking interleukin-1 in rheumatic diseases. Ann N Y Acad Sci. 2009 Dec;1182:111-23.
- CLUSTER ClinicalTrials.gov, URL: https://clinicaltrials.gov/ct2/show/NCT02059291 (актуализация на 26.07.2024 г.).
- Ter Haar N.M. et al. Ann Rheum Dis 2015;0:1–9. doi: 10.1136/annrheumdis-2015-207546.
Оцените материал:


